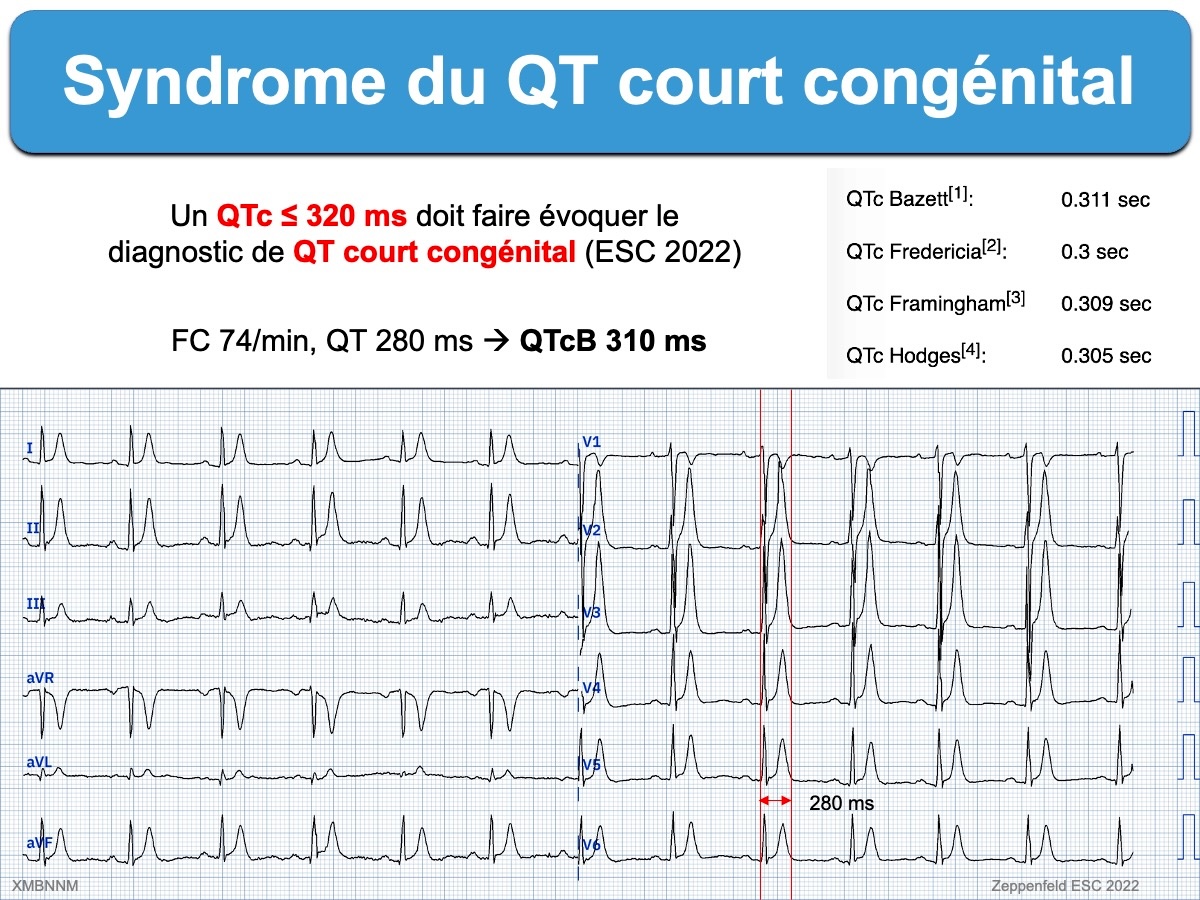
Syndrome du QT court congénital 6 novembre 202318 novembre 2019 par Pierre Taboulet Voir Intervalle QT court