« Maladie du myocarde (cf. Cardiomyopathie) qui entraine une hypertrophie ventriculaire gauche et/ou droite ou seulement une portion » (AHA 2020) [1], (ESC 2014) [2]. C’est une maladie génétique autosomique dominante à pénétrance variable et qui touche 0,2-0,5% de la population [1][4].
Le diagnostic est posé en général après 35 ans, parfois plus tôt (6% < 16 ans [12]). Il est parfois évoqué lors de la réalisation systématique d’un ECG (ex. Certificat de non-contrindication au sport) ou, plus souvent, à l’occasion de complications cliniques (insuffisance cardiaque, syncope, dyspnée) ou électriques : anomalies de conduction (bloc AV ou bloc de branche), anomalies du rythme supraventriculaire (dysfonction sinusale, FA…) ou ventriculaire (ESV ou tachycardies ventriculaires).
L’ECG est évocateur dans la majorité des cas, l’échographie est plus précise, mais le gold standard est l’IRM. Il est exceptionnellement normal (AHA 2000 [1] ESC 2014 [2] et revue 2023 [14]).
Le diagnostic différentiel principal est une hypertrophie ventriculaire secondaire à une adaptation (ex. sport, HTA, obstacle à l’éjection), mais d’autres hypothèses doivent être éliminées. Il est recommandé de prendre l’avis d’un centre expert, en cas de doute ou pour le diagnostic précis, le dépistage familial et la prise en charge.
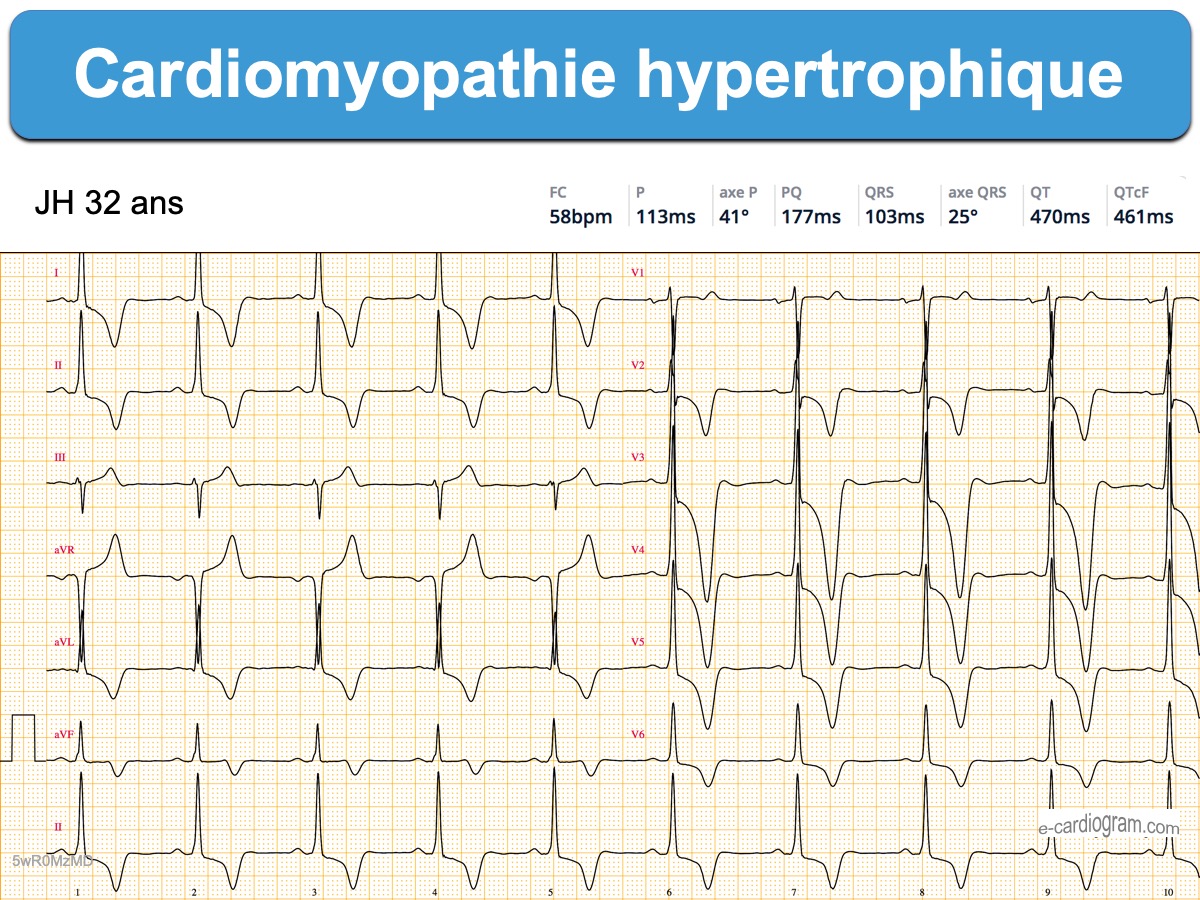
Le pattern ECG d’une CMH peut être typique
Les signes électriques directs ou indirects varient selon le phénotype [5]. Les deux critères ci-dessous sont relativement constants.
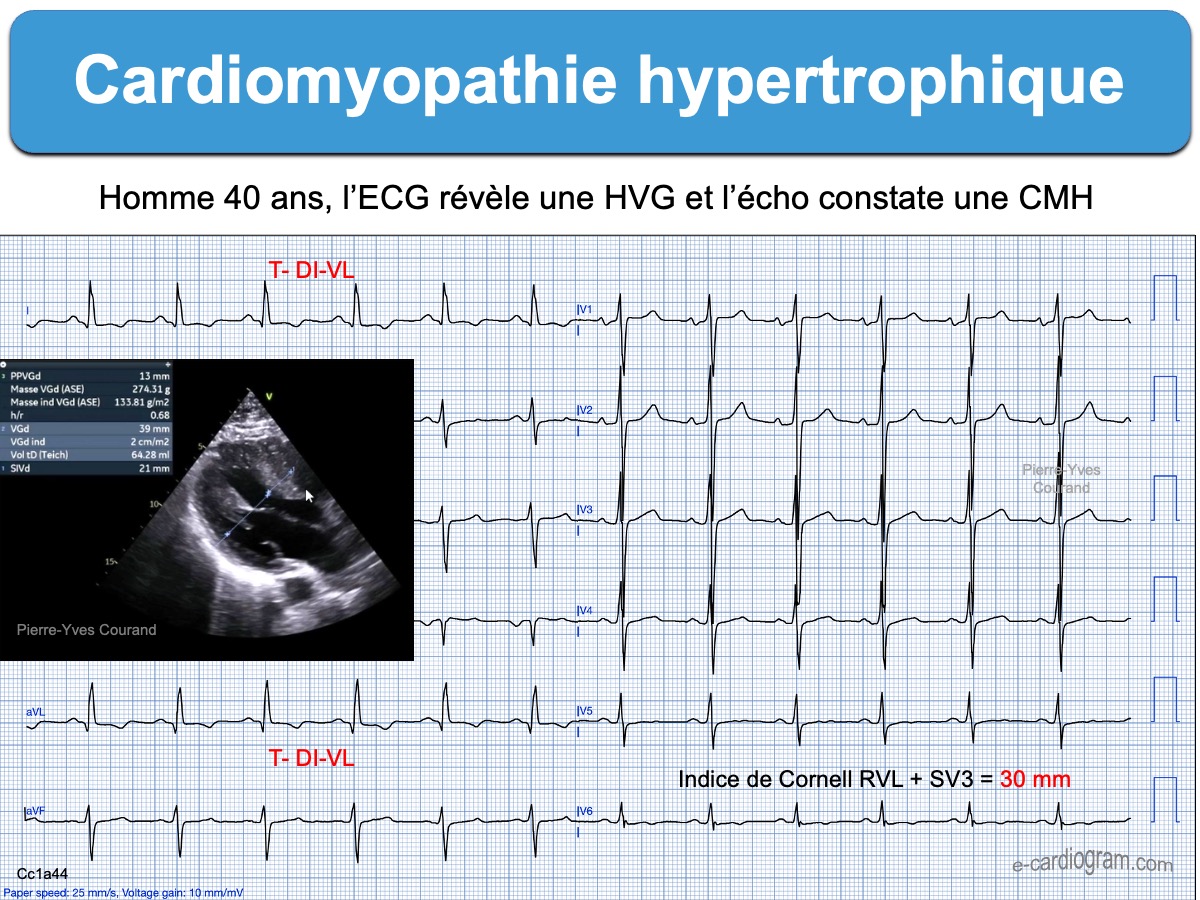
- Ondes R et/ou ondes S anormalement amples de V2 à V6 (et souvent DI-VL), avec élargissement de l’onde R (« retard de la déflexion intrinsécoïde« ) en rapport avec l’augmentation de la masse musculaire ventriculaire gauche. En cas de négativité (fréquente) de l’indice de Sokolow, il faut mesurer d’autres indices d’hypertrophie VG).
- Sous-décalage de ST ou/et surtout des ondes T inversées géantes (> 10 mm ou 1 mV) dans les dérivations inférolatérales qui ne comportent pas d’onde Q (forme apicale) [8].
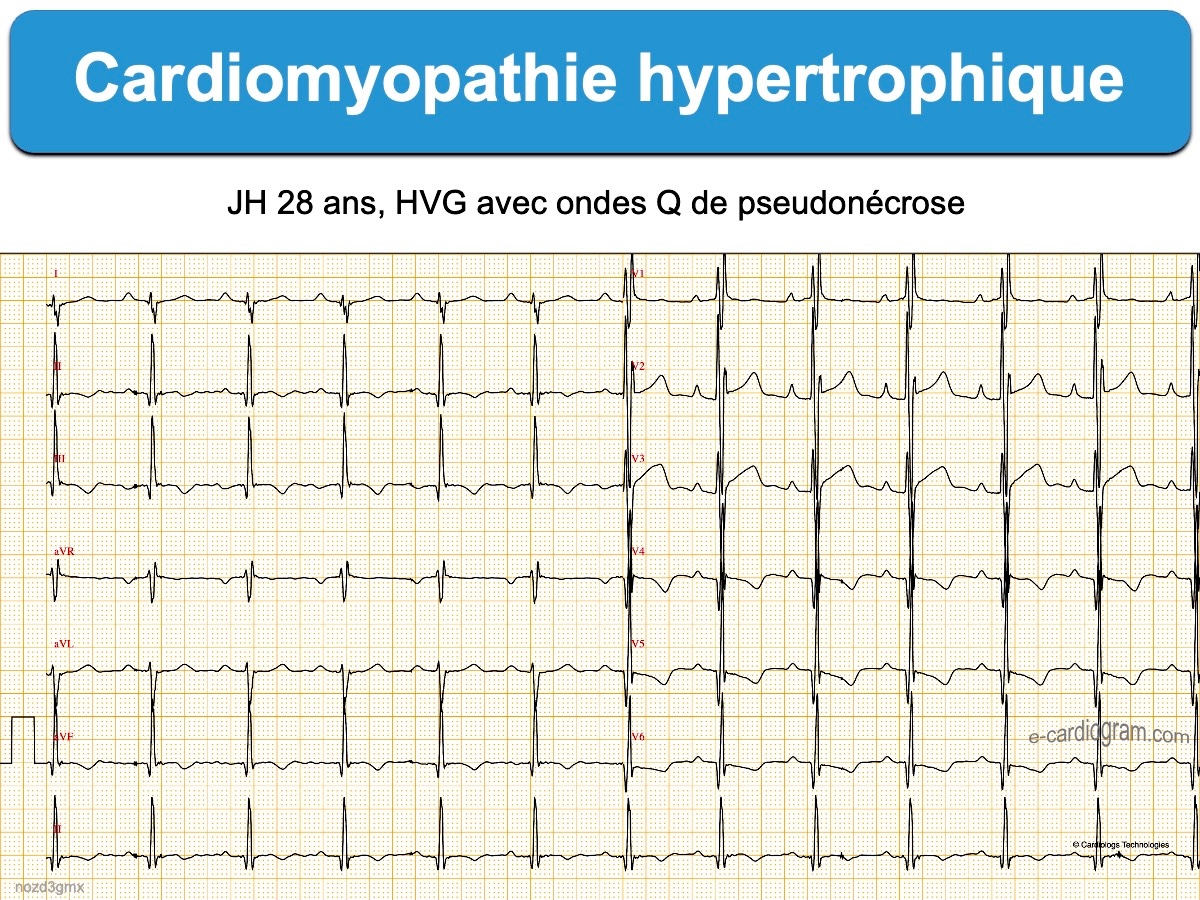
- Des ondes Q anormales, dites ondes Q de pseudo-nécrose, sont inconstantes mais hautement évocatrices, Elles sont souvent plus fines que dans la séquelle de nécrose et précédent des ondes R amples au début de l’évolution (voir exemple [7]). Elles sont dues à un remplacement du muscle par de la fibrose ou un vecteur initial de dépolarisation opposé à l’électrode ou la dérivation qui enregistre.
- Un intervalle P-R court est possible, mais une vraie préexcitation ventriculaire s’observe plutôt dans les maladies de surcharge type maladie de Danon (déficit en LAMP2, liée à l’X) ou de Pompe infantile [10][11].
- Des QRS microvoltés en V6-DI-VL sont possibles en fin d’évolution.
- Des ondes T positives amples associées à des ondes Q sont possibles (forme septale). Des ondes T diphasiques ou peu amples sont possibles.
- Un sus-décalage de ST anormal est possible, peu ample, plutôt ascendant, prolongé par des ondes T inversées [9]. Il peut être confondant, dans certaines dérivations, avec un infarctus ST+ (voir ECG ci-dessous).
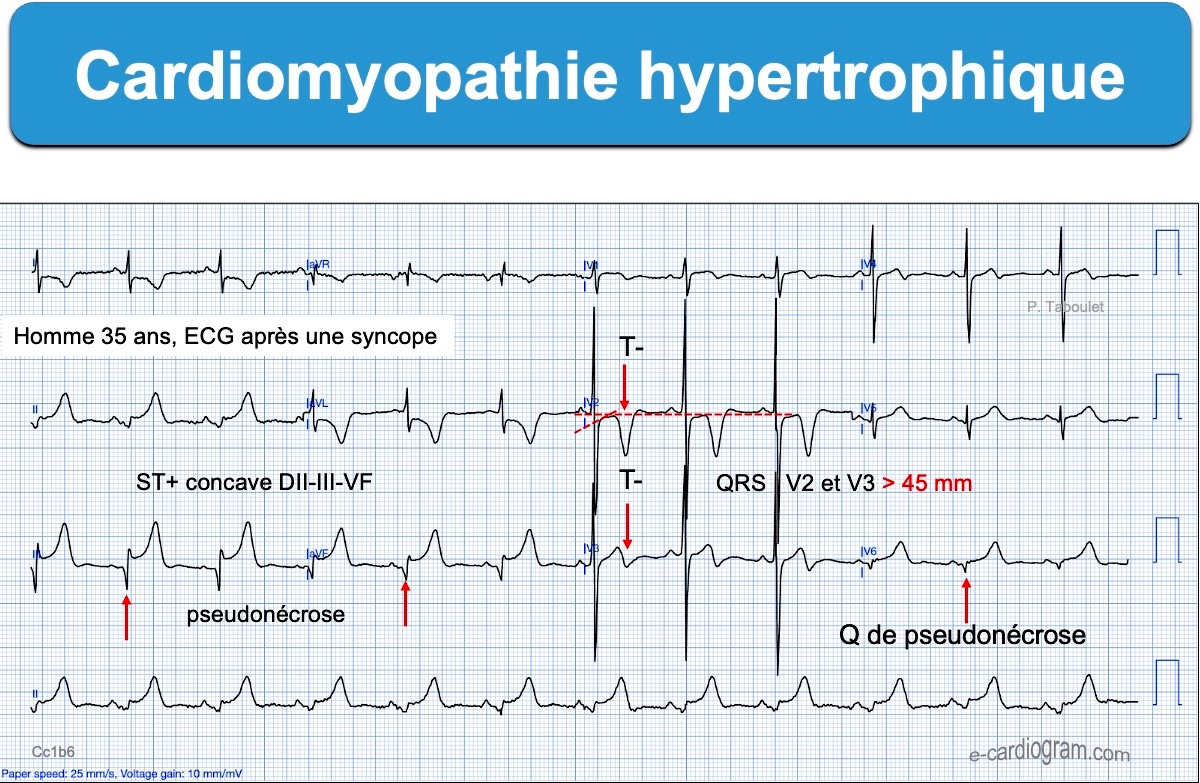
- Certaines CMH ont des QRS tellement amples qu’ils ne « tiennent » pas dans le format de l’ECG, aussi une réduction de calibrage est nécessaire, parfois automatiquement réalisée par l’électrocardiographe.
- D’autres CMH se présentent sous la forme d’une HVG banale, ou pire, des QRS non amples mais des ondes T anormales. Il faut donc y penser +++
La cardiomyopathie hypertrophique apicale
C’est une forme rare de cardiomyopathie hypertrophique qui prédomine chez les patients asiatiques (25% des CMH au japon vs 2 à 9% en occident) (Sakamoto 1976, Yamaguchi syndrome [8]).
- L’ECG est évocateur en cas d’ondes T inversées géantes (souvent > 10 mm ou 1 mV) dans les dérivations précordiales ou inféro-latérales, en regard de grandes ondes R discrètement élargies. Dans ces dérivations, le segment ST débute souvent en dessous de la ligne de base, puis il est ascendant, à l’inverse des HVG de surcharge volumétrique (HTA rétrécissement aortique…). L’onde T inversée se termine légèrement positivement.
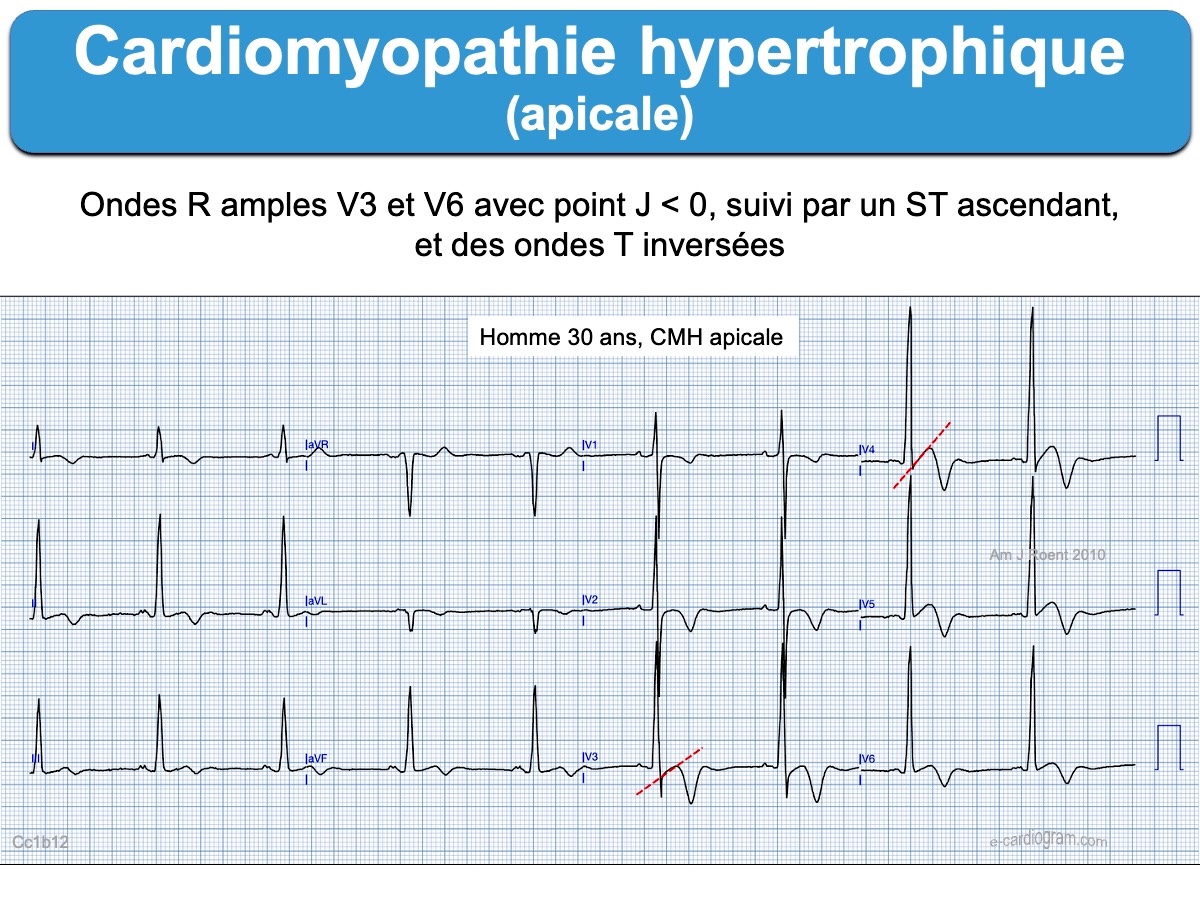
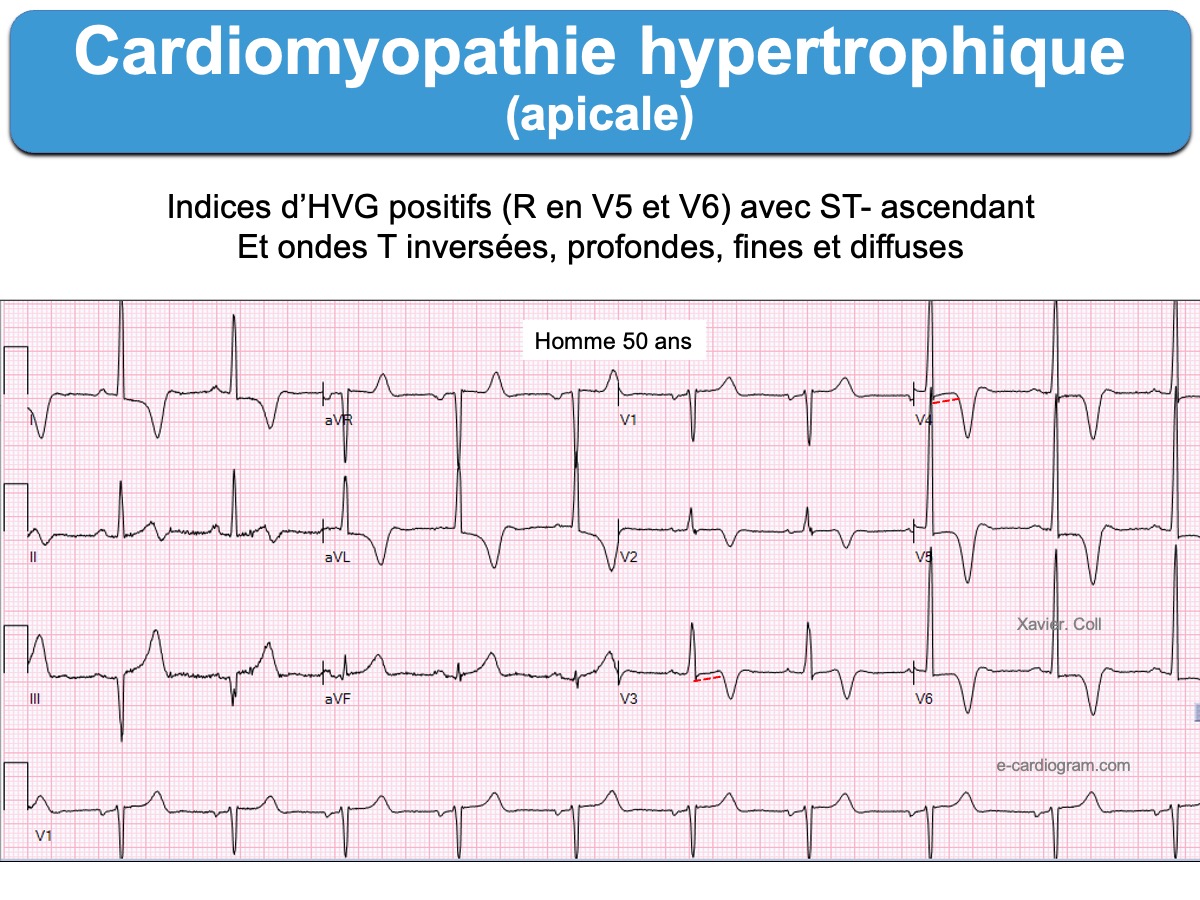
- Elles n’entrainent pas d’obstruction dynamique à l’éjection du VG.
- Voir IRM d’une CMH apicale (superbe) ici
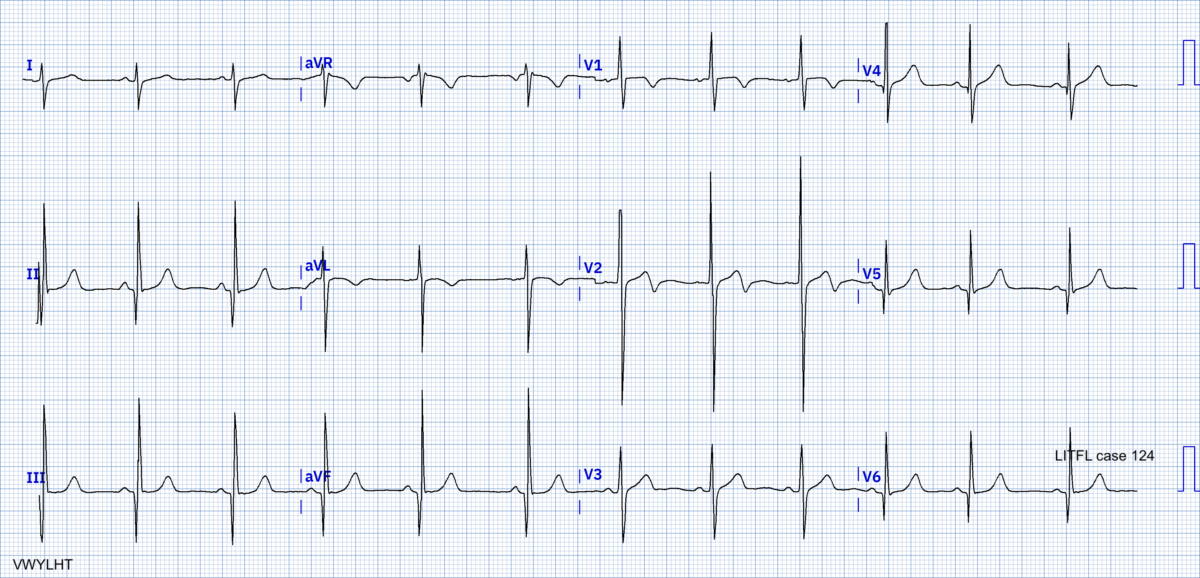
La cardiomyopathie hypertrophique septale
- L’ECG peut être évocateur devant l’existence d’ondes Q profondes, mais peu larges (Q de pseudo-nécrose) dans les dérivations précordiales en regard de grandes ondes R ou non. Une grande onde R initiale en V1-V2-V3 (qui explorent le septum) peut traduire l’hypertrophie septale.
- Elles entrainent une obstruction dynamique à l’éjection du VG.
- Voir cas clinique avec écho cœur LITFL [7]
Diagnostics différentiels
- Hypertrophie ventriculaire secondaire à une adaptation (ex. sport, HTA, obstacle à l’éjection comme un RAC)
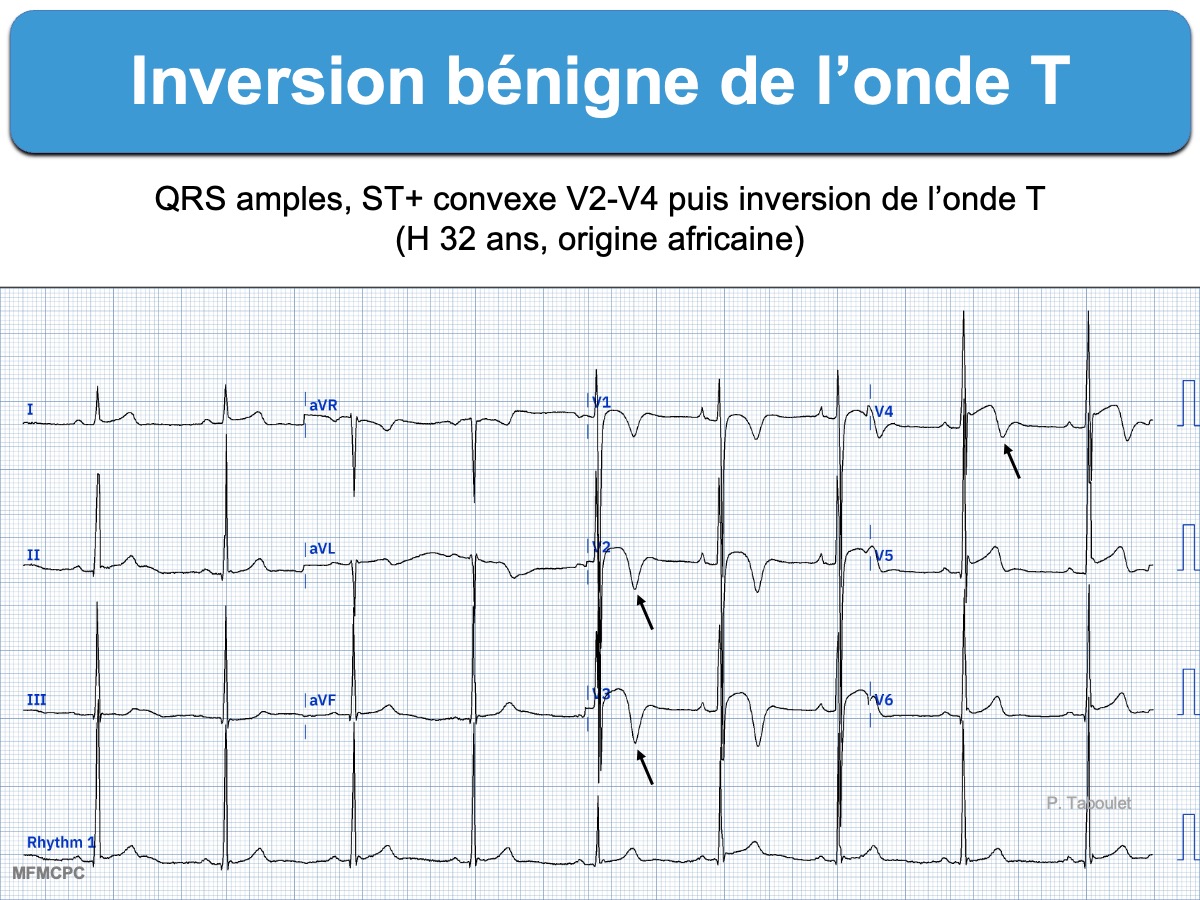
- Variante de repolarisation normale chez les sujets d’origine africaine et les athlètes (cf. Black repolarisation) [10]. L’onde T s’inverse à partir d’un segment ST ascendant, qui démarre initialement au-dessus de la ligne de base (≠ CMH).
- Amylose cardiaque : en cas de microvoltage diffus
- Maladie de Fabry
- Maladie de Pompe infantile (glycogénose de type II, déficit en α-glucosidase)
Complications
- Syncope, insuffisance cardiaque, fibrillation atriale, arythmie ventriculaire
- La mort subite est plus fréquente chez l’enfant et l’adulte jeune (7 à 8 %/an), annoncée par des facteurs de risque [6]. Elle est parfois le mode de révélation (âge moyen 40 ans, 14-57 ans) [13]. Chez les patients à haut risque, la pose d’un défibrillateur implantable automatique est recommandée de façon prophylactique (cf. Recommandations ESC 2021 [2]).
Traitement
Le traitement des symptômes invalidant repose sur les médicaments du type bêtabloquant ou inhibiteur calcique, proposés pour diminuer la contractilité du muscle cardiaque, mais parfois insuffisamment efficaces. L’adjonction de diurétique est souvent nécessaire prudemment pour diminuer les signes congestifs.
Plus rarement, on fait appel à des solutions plus invasives, comme l’alcoolisation septale (injection d’alcool dans la coronaire) ou la chirurgie (myomectomie). Mais il existe de nouvelles molécules prometteuses, comme les inhibiteurs sélectifs de la myosine cardiaque. Les indications doivent être posées par des spécialistes dans des centres de références spécialisés.
Cas cliniques
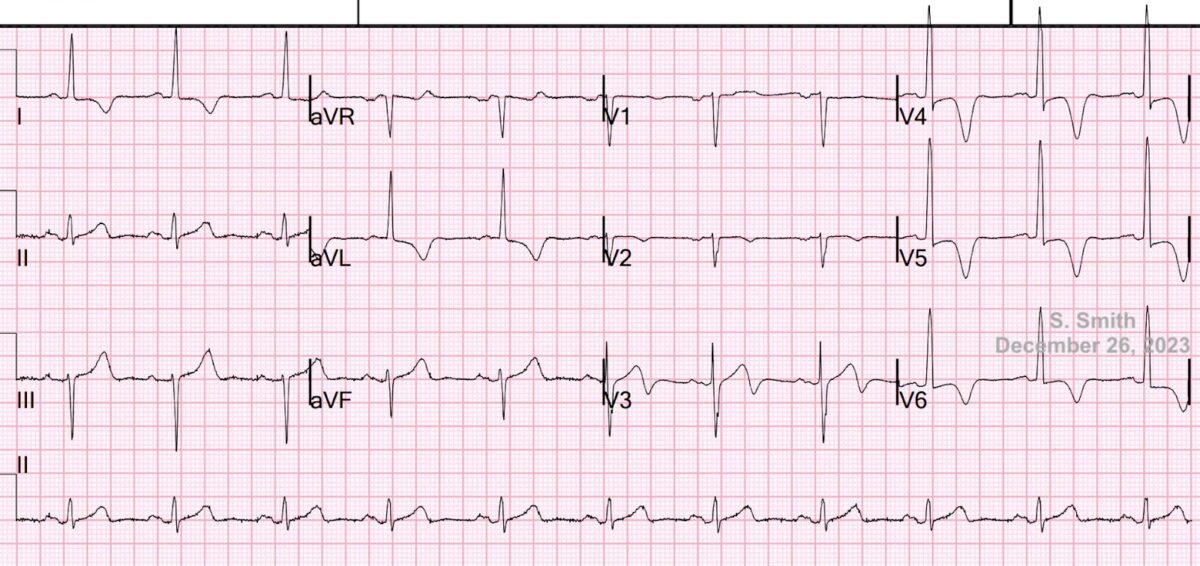
Cas clinique de S. Smith (ECG archi typique). An elderly patient with stuttering chest pain. Don’t jump to conclusions
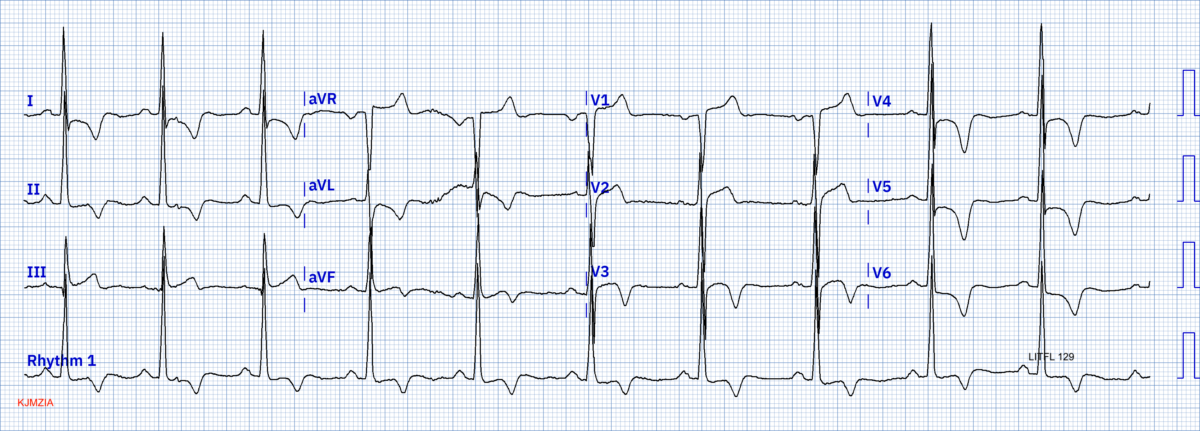
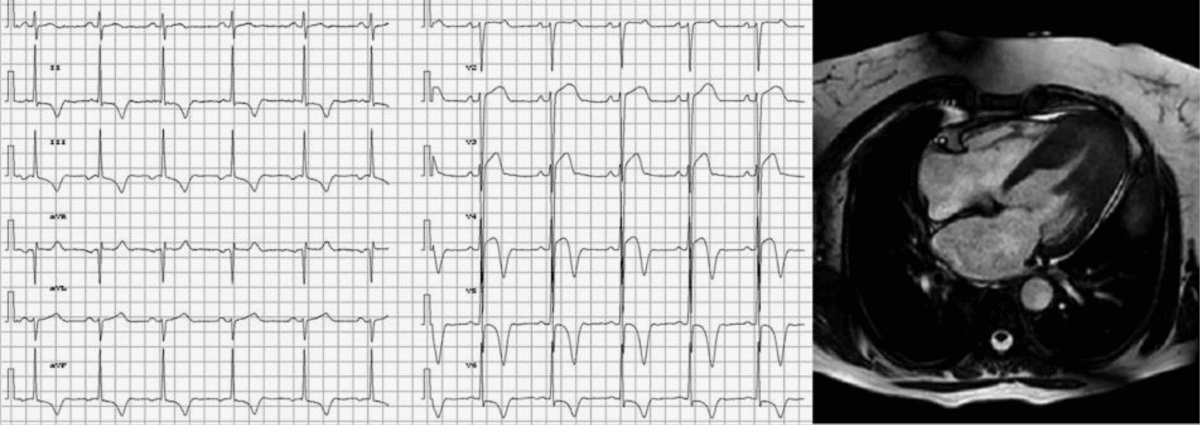
Cas cliniques Bernardini [14]
Guidelines
Guidelines [ESC 2014] [2]
Guidelines AHA/ACC 2020 [1]
Congrès marocain : histoire familiale de CMH très bien documentée (MC Malergue 2021).
Références (abonnés)
La suite est réservée aux membres et stagiaires du site.
Se connecter | Devenir membre | Devenir stagiaire