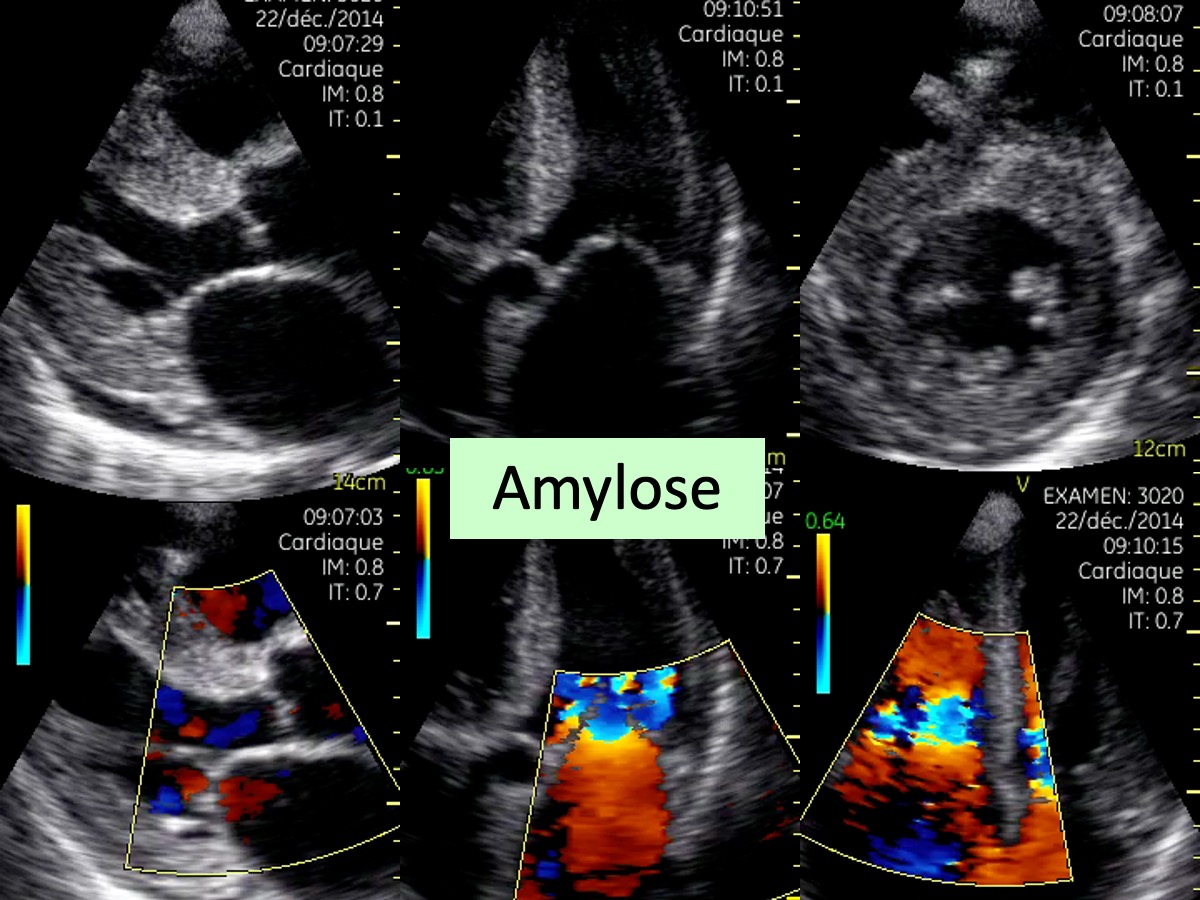
Infiltration pariétale du cœur par des dépôts amyloïdes. Cette infiltration peut affecter toutes les propriétés électriques et contractiles du muscle cardiaque.
Synonymes : Cardiomyopathie ou cardiopathie amyloïde
Le diagnostic est évoqué devant
- des signes cliniques extra-cardiologiques (ex. neuropathie périphérique ou SNA, paresthésies, macroglossie, canal carpien ou Dupuytren, atteinte rénale…) qui font rechercher une atteinte cardiaque
- ou devant des signes cardiologiques (ex. hypertrophie ventriculaire sévère en échocardiographie ou pattern ECG) qui font rechercher une amylose.
Signes ECG
On peut évoquer une amylose devant un pattern typique associant un microvoltage, un rabotage des ondes R de V1 à V4 et/ou des ondes q de pseudo-nécrose (« pseudo-infarct pattern« ). Ces signes manquent 1 fois sur 2 [1][2][3][4]. Fait notable, ces signes sont discordants avec l’échocardiographie qui montre des signes francs d’hypertrophie ventriculaire.
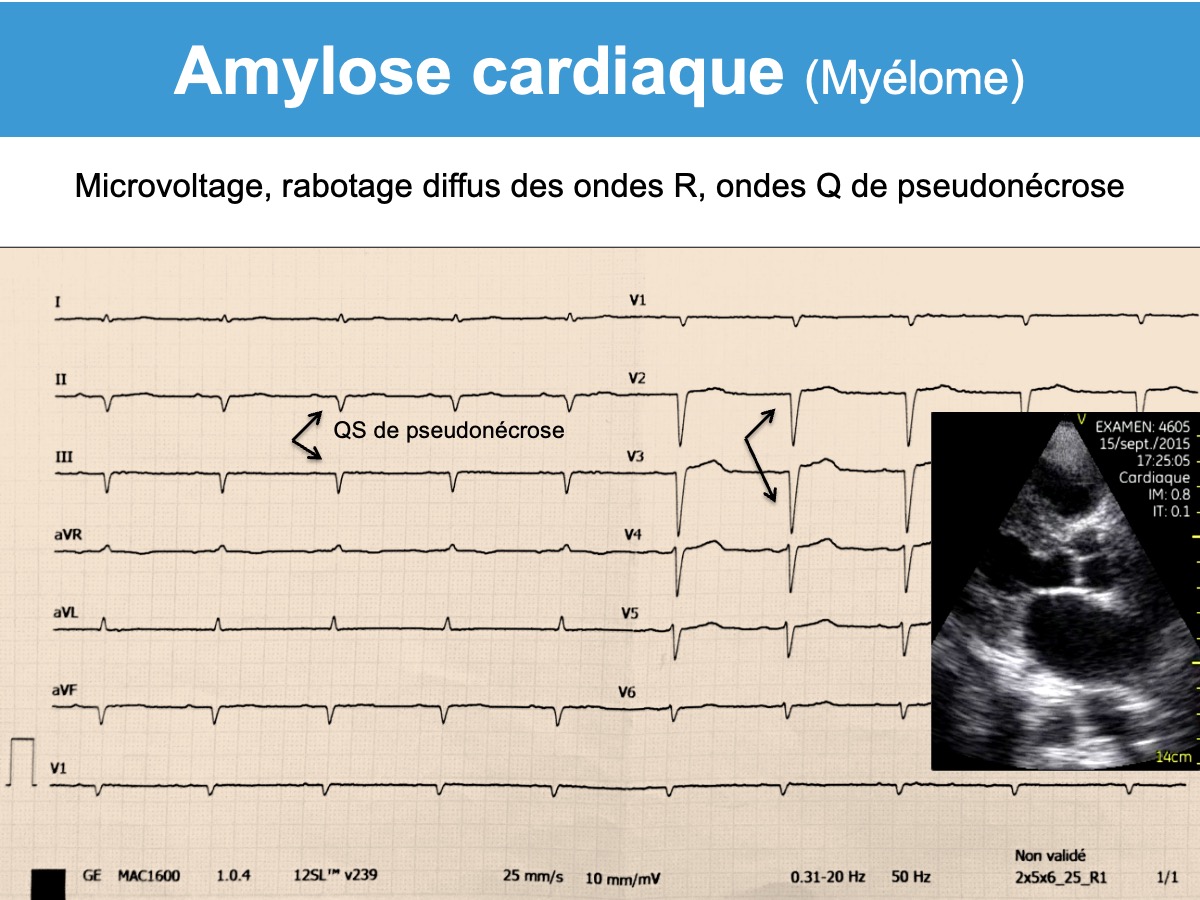
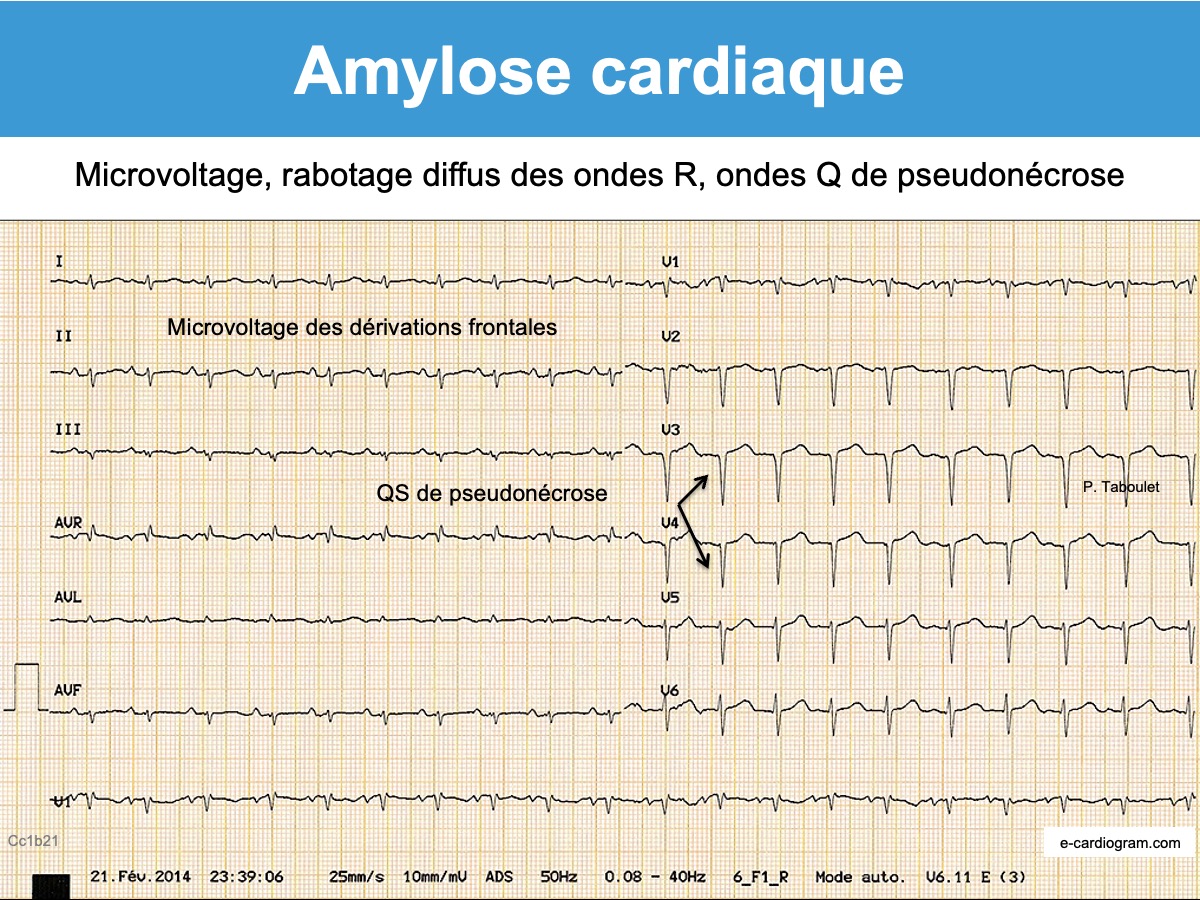
D’autres signes sont fréquents au cours de l’évolution mais non spécifiques [1][2][3]:
- des ondes P peu voltées ou dysmorphiques et parfois bloc interatrial ou bloc sinoatrial ou dysfonction sinusale
- anomalies du rythme atrial (FA, flutter atypique…)
- bloc AV
- bloc de branche (surtout BBG) ou bloc intraventriculaire
- anomalies non spécifiques de la repolarisation
- et plus rarement arythmie ventriculaire.
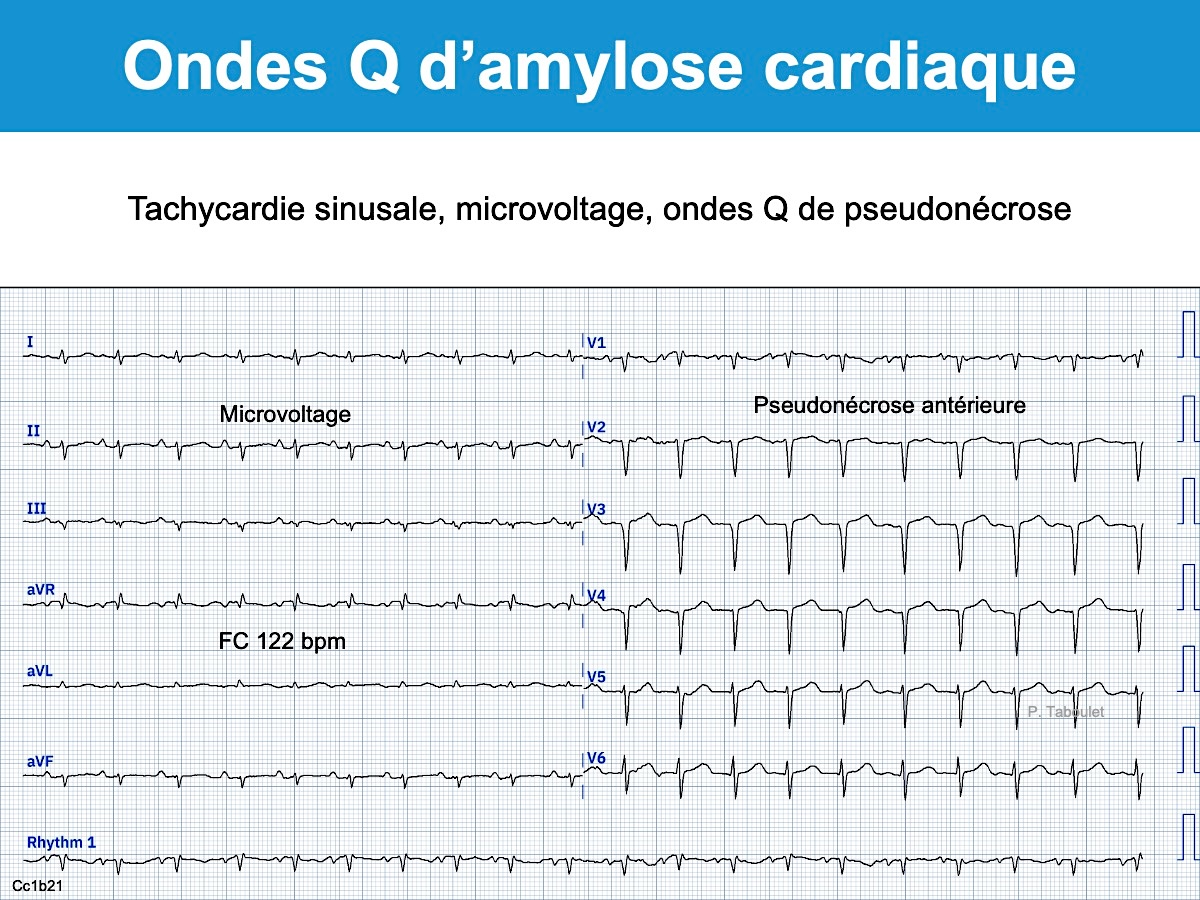
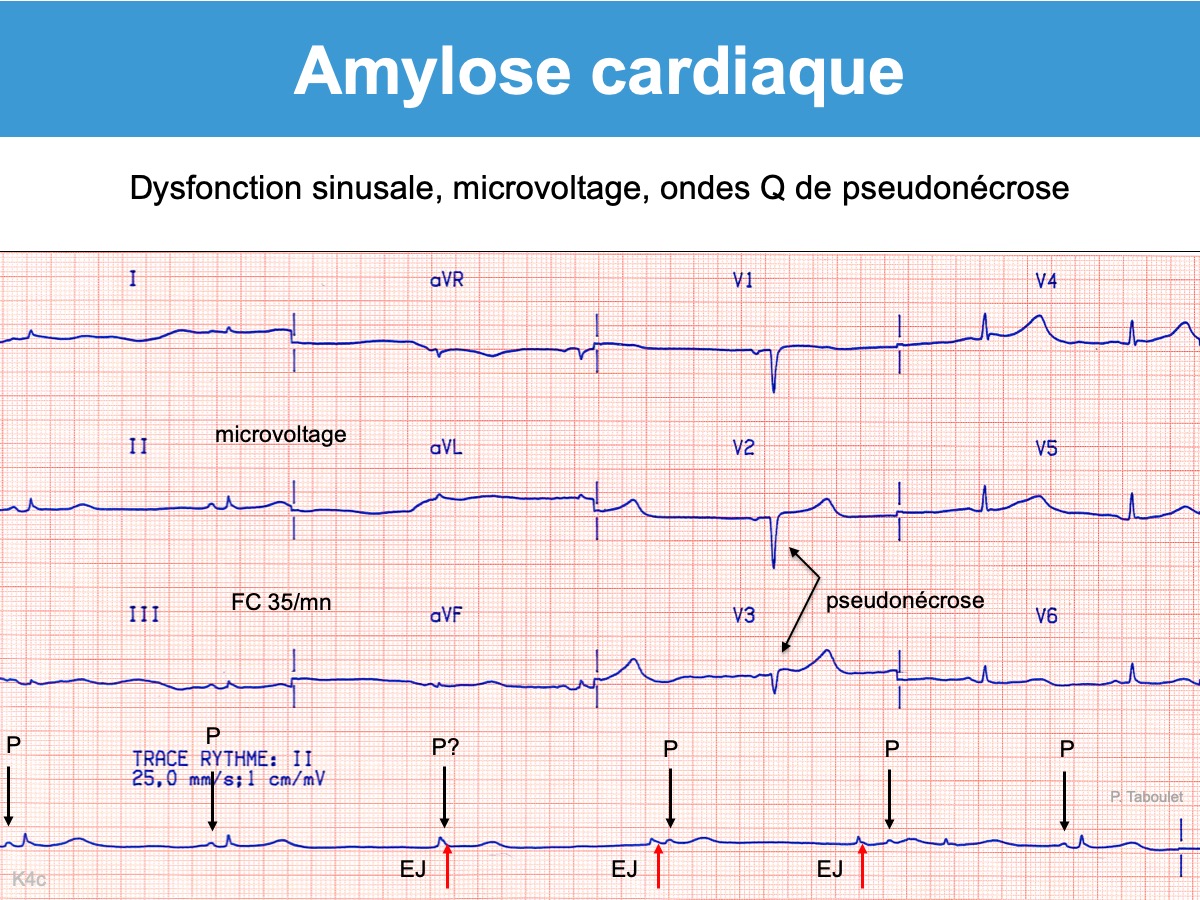
L’échocardiographie (hypertrophie ventriculaire) et un tracé ECG caractéristique (microvoltage, pseudonécrose) doivent faire suspecter une amylose cardiaque [1].
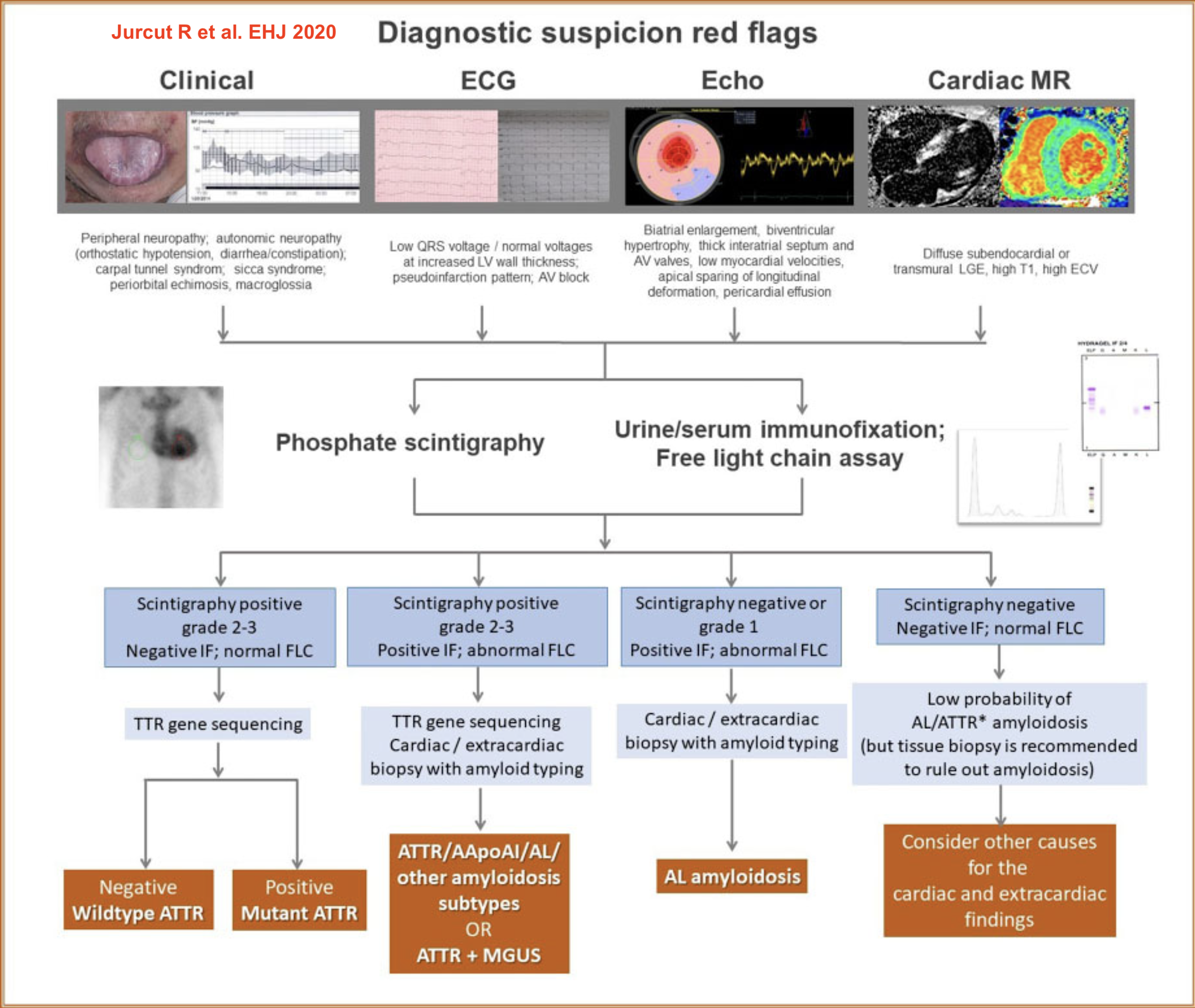
Le diagnostic étiologique repose en première intention sur la clinique et éventuellement une IRM avec rehaussement tardif (pour les diagnostics différentiels) [4][5].
En deuxième intention, une électrophorèse avec immunofixation des protéines (S et U) à la recherche d’une gammapathie monoclonale (light-chain amyloid AL) et une scintigraphie osseuse aux diphosphonates (à la recherche d’une amylose TTR diffuse),
D’autres examens sont parfois utiles comme une histologie de la glande salivaire ou d’un autre organe (dépôts d’amylose rouge Congo, biréfringence) et des tests génétiques en cas de cardiomyopathie amyloïde familiale (pour la distinguer d’une amylose sénile…).
La cardiomyopathie amyloïde familiale liée à la transthyrétine (TTR) est une amylose systémique héréditaire liée à la TTR (ATTR) avec des manifestations cardiaques prépondérantes (cardiomyopathie restrictive avec divers degrés d’insuffisance cardiaque chronique et de possibles brady/tachyarrhythmies), dues à des infiltrations myocardiaques d’une protéine amyloïde anormale [1].
Diagnostic et traitement (ESC 2021 [5])
Principes thérapeutiques CV : diurétiques, anticoagulant (parfois même en RS si contraste spontané et risque hémorragique faible), antiarythmique fréquemment nécessaire à l’étage atrial (amiodarone, ablation de FA à envisager), évolution vers pacemaker fréquente. Ni bêtabloquant ni IEC car très mal tolérés.
Si vous souhaitez améliorer cette page, contactez-moi, merci
Cas cliniques Cardio-online
Réseau Amylose CHU Mondor – Paris

Si vous souhaitez améliorer ce contenu, merci de me contacter
Livre ECG de A à Z et autres (P. Taboulet, 2e ed. 2025)
Faîtes des quiz (site web)
YouTube : ma playlist https://www.youtube.com/c/PierreTaboulet-ECG)
YouTube : ECG Minute (hebdomadaire 10 min)
Inscrivez-vous à ma newsletter hebdomadaire (https://www.e-cardiogram.com/newsletter)
Amylose cardiaque à transthyrétine sauvage et références (réservées aux abonnés)
La suite est réservée aux membres et stagiaires du site.
Se connecter | Devenir membre | Devenir stagiaire